Kanzerogenese des Prostatakarzinoms
Prostatakrebsrisiko = [(Familiäre Vorbelastung + Androgenrezeptor- und Östrogenrezeptor-alpha-stimulierende Hormone und Fremdöstrogene + Kanzerogene + IGF-1 + Insulin + Übergewicht + chronische Prostatitis) x Jahre der Exposition] – [(Östrogenrezeptor-beta-stimulierende Hormone und Pflanzenstoffe + Krebshemmstoffe + Bewegung) x Jahre der Exposition]
Diese Gleichung lässt sich auch in einer Abbildung darstellen (Abb. 1):
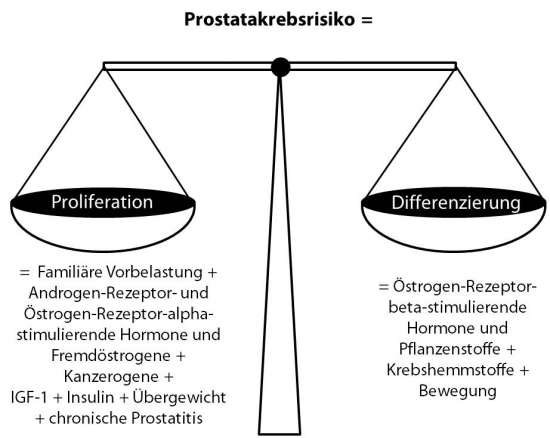
Abb. 1: Einflussfaktoren auf das Gleichgewicht aus Proliferation und Differenzierung, das sich entscheidend auf das Prostatakrebsrisiko auswirkt
Dreistufenmodell der Kanzerogenese
Das klassische Dreistufenmodell der Kanzerogenese beschreibt die Tumorentwicklung in den Abschnitten Tumorinitiation, -promotion und -progression. Auch wenn dieses Erklärungsmodell inzwischen durch das komplexere Mehrschrittmodell abgelöst wurde (Harris, 1991), hat es sich dennoch aufgrund seiner einfachen Darstellung etabliert.
Bei der Initiation, dem ersten Schritt der Kanzerogenese, erfährt die Zelle eine durch ein Kanzerogen (z. B. freie oxidierende Radikale (ROS), Alkylantien, Chinone (aus der Phase-1-Metabolisierung von Östrogenen), Entzündungsprozesse, Steroidhormone) ausgelöste Mutation. Diese kann nur dann bestehen bleiben, wenn sie nicht durch DNA-Reparaturenzyme beseitigt wird oder die Zelle nicht in die Apoptose getrieben wird.
In der Promotionsphase erfahren die initiierten Zellen über Jahre oder Jahrzehnte hinweg einen Wachstumsstimulus. Hormone (Androgene, Östrogene), Entzündungsprozesse und freie Radikale spielen als Promotoren des Prostatakarzinoms eine tragende Rolle.
Die Progression stellt schließlich den irreversiblen Übergang vom gutartigen Tumor (d. h. der präneoplastischen Läsion) zum bösartigen (d. h. invasiven und metastasierenden) Tumor dar. Charakterisiert wird die Tumorprogression durch weitere genetische Schäden, die Aktivierung von Proto-Onkogenen und die Inaktivierung von Tumorsuppressor-Genen. Die Fähigkeit des Tumors zur Invasion und Metastasierung kennzeichnet die dritte Phase der Kanzerogenese, wobei deren Ausmaß für die Morbidität und Letalität von Tumorpatienten entscheidend ist. Der Begriff Tumorzellinvasion beschreibt dabei die Fähigkeit der Zellen zur Überquerung anatomischer Barrieren (z. B. Basalmembranen, interstitielles Stroma, Interzellularbrücken). Die weitere Progression von einem invasiven zu einem metastasierenden Karzinom ist durch das Eintreten von Krebszellen in und das Austreten derselben aus einem Blut- oder Lymphgefäß charakterisiert, gefolgt von der Invasion in ein sekundäres Gewebe.
Da das Prostatakarzinom ein typischer, fast immer hormonabhängiger Alterskrebs ist, spielt für das individuelle Risiko vor allem die Tumorpromotion eine entscheidende Rolle. Der Hormon- und Rezeptorstatus des Tumorgewebes sowie die Belastung und Belastbarkeit durch oxidativen Stress und Entzündungsprozesse beeinflussen dabei maßgeblich das Schicksal des Patienten. Die individuelle Situation wird sowohl durch die genetische Disposition als auch durch die Ernährungs- und Lebensweise bedingt.
Der Begriff Prostatakarzinom umfasst eine Vielzahl von unterschiedlichen klinischen Verläufen. Diese Vielfalt erklärt sich durch unterschiedlich differenzierte Zelltypen, aus denen sich ein Prostatakarzinom entwickeln kann und die sich bei der Entwicklung des Karzinoms von differenzierten, Androgen-sensiblen zu immer undifferenzierteren, Androgen-resistenten oder scheinbar Androgen-unabhängigen Zelltypen verändern.
Zellbiologie des Prostataepithels
Zellbiologisch lassen sich im Prostataepithel drei Zelltypen unterscheiden (Bonkhoff und Remberger, 1996; Foster et al., 2000):
Die PSA-produzierenden, sekretorischen Zellen bilden die Hauptmasse des Prostataepithels und das Differenzierungskompartiment. Sie tragen den Androgenrezeptor (AR) und sind Androgen-abhängig, haben jedoch eine geringe Proliferationskapazität. Unter Androgenentzug erleiden die sekretorischen Zellen den programmierten Zelltod und sterben ab.
Die Basalzellen bilden das Stammzell- und Proliferationskompartiment. Sie grenzen das sekretorische Epithel vom Stroma (Bindegewebe der Prostata) ab und erhalten eine normale Beziehung zwischen dem Prostataepithel und dem Stroma aufrecht. Die Basalzellschicht enthält die teilungsfähigen Zellen, aus denen sich das Prostataepithel regeneriert. Die Basalzellen sind zwar Androgen-unabhängig, einige Basalzellen exprimieren jedoch den AR. Aus diesen AR-positiven Basalzellen entstehen die sekretorischen Zellen, die immer AR-positiv sind. Die Basalzellen sind resistent gegenüber dem Androgen-regulierten, programmierten Zelltod.
Die neuroendokrinen Zellen kommen verstreut im Prostataepithel vor. Sie sind extrem langlebig und teilen sich nicht mehr. Sie tragen keinen AR und exprimieren den endokrinen Marker Chromogranin A.
Die Differenzierungsvorgänge zwischen den verschiedenen Zelltypen und funktionellen Kompartimenten, die letztlich die Integrität des Prostataepithels aufrechterhalten, werden durch ein hormonelles Gleichgewicht zwischen Androgenen und Östrogenen bestimmt:
Die Differenzierung einer Basalzelle zu einer sekretorischen Zelle ist ein Androgen-regulierter Prozess und abhängig von der Anzahl der AR-positiven Basalzellen, die in diesen Differenzierungsprozess eintreten. Demgegenüber stehen die Östrogene, die diesen Differenzierungswandel blockieren und somit zur Verkümmerung des sekretorischen Epithels und zur Basalzellhyperplasie führen. Dieser Östrogeneffekt wird über den klassischen Östrogenrezeptor alpha (ER-alpha) vermittelt, der in der Basalzellschicht des Prostataepithels exprimiert wird. Im Stroma der Prostata werden Östrogen- und ER-alpha-vermittelt Wachstumsfaktoren gebildet, welche die Proliferation des Prostataepithels anregen. Östrogene stimulieren somit die Proliferation und hemmen die Zelldifferenzierung.
Der 1996 von Gustafsson entdeckte ER-beta, der vorzugsweise pflanzliche Phytoöstrogene bindet, wird dagegen überwiegend im sekretorischen Epithel exprimiert. Genetisch manipulierte Mäuse, denen der ER-beta fehlt, entwickeln im Alter spontan eine gutartige Prostatahyperplasie (Krege et al., 1998; Weihua et al., 2001; Bonkhoff et al., 2003). Demnach schützt ein funktioneller ER-beta das Prostataepithel der Maus vor Hyperplasie.
Prämaligne Transformation: Prostatische intraepitheliale Neoplasie
Die prämalignen Vorläufer des Prostatakarzinoms werden unter dem Begriff „Prostatische intraepitheliale Neoplasie“ (PIN) zusammengefasst (Foster et al., 2000). Dabei wird zwischen Iow grade (LGPIN) und high grade (HGPIN) PIN unterschieden. Eine HGPIN ist Vorläufer von Prostatakarzinomen der Kategorie Gleason ≥ 7, während die atypische adenomatöse Hyperplasie ein möglicher Vorläufer hochdifferenzierter Prostatakarzinome (Gleason 6) ist. Nach Autopsiestudien geht HGPIN dem Auftreten eines Prostatakarzinoms um etwa 10 Jahre voraus (Wu et al., 2004).
Bei der bösartigen Transformation des Prostataepithels (HGPIN) kommt es zu schweren Differenzierungs- und Proliferationsstörungen im Zellsystem des Prostataepithels (Foster et al., 2000). Die Proliferationsaktivität verlagert sich aus der Basalzellschicht (Proliferationskompartiment) in das sekretorische Epithel (Differenzierungskompartiment). Diese Umverteilung der Proliferationszone beruht auf einer abnormen Expression von Steroidrezeptoren (AR, ER-alpha, ER-beta) und Wachstumsfaktorrezeptoren (HER-1, HER-2, HER-3), die im normalen Prostataepithel im Proliferationskompartiment exprimiert und deren Liganden im Prostatastroma produziert werden (Bonkhoff et al., 1998).
Hinzu kommen Störungen in der Regulierung des programmierten Zelltodes: Bcl-2, das im normalen Prostataepithel die Basalzellschicht vor dem programmierten Zelltod schützt, wird in etwa 20 % der HGPIN im sekretorischen Epithel überexprimiert und verhindert so im transformierten sekretorischen Epithel den programmierten Zelltod. Bcl-2-positive HGPIN zeigen auch eine verminderte oder fehlende AR-Expression, wodurch ihre Androgensensitivität abgesenkt oder sogar eingebüßt wird (Bonkhoff et al., 1998 und 2003).
Einfluss der Hormone auf die Kanzerogenese
Altersbedingte hormonelle Veränderungen
Verschiebung des Östrogen-/Androgengleichgewichts
Der Prostatakrebs ist ein typischer Alterskrebs: Seine höchste Inzidenz liegt jenseits des 65. Lebensjahres. Auf den ersten Blick ist es erstaunlich, dass das Androgen-abhängige Prostatakarzinom seine höchste Inzidenz zu dem Zeitpunkt im Leben des Mannes hat, an dem der Androgeneinfluss am niedrigsten und der Östrogeneinfluss am höchsten ist. Dies weist auf die besondere Bedeutung der Östrogene in der Kanzerogenese des Prostatakarzinoms hin.
Mit zunehmendem Alter verschiebt sich bei Männern das Androgen-/Östrogenverhältnis auf die Seite der Östrogene. Diese Verschiebung des Hormongleichgewichts geschieht durch eine Abnahme der Testosteron- und Dehydroepiandrosteron- (DHEA-)Produktion. Gleichzeitig werden aus Testosteron immer mehr Östrogene und immer weniger Dihydrotestosteron (DHT) gebildet. Mit dem Alter nehmen so die Östrogenkonzentrationen, die den ER-alpha aktivieren zu, während die Spiegel der typischen ER-beta-Agonisten (3beta-Adiol, DHEA) abnehmen (s. Abb. 2). In den folgenden Abschnitten werden die komplexen Gleichgewichte von Östrogenen bzw. Androgenen und deren Rezeptoren diskutiert, die in ihrem fein abgestimmten Zusammenspiel die Entstehung des Prostatakarzinoms maßgeblich beeinflussen.
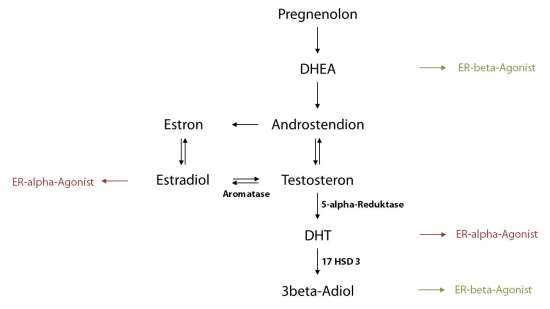
Abb. 2: Bildungswege und ER-Bindungspräferenzen verschiedener Androgen- und Östrogenmetaboliten
Auch bei der Entwicklung der gutartigen Prostatahyperplasie steht der relative Androgenmangel mit der altersbedingten Verschiebung des Androgen-/Östrogengleichgewichts im Vordergrund (Bonkhoff und Remberger, 1998): Der relative Androgenmangel führt zur Überexpression des AR im Proliferationskompartiment (hypersensitive Basalzellen) und zur Beschleunigung des Differenzierungswandels von Basalzellen zum sekretorischen Zelltyp mit einer Hyperplasie des sekretorischen Epithels. Darüber hinaus verursacht der relative Androgenmangel eine verminderte Expression des Androgen-regulierten ER-beta im sekretorischen Epithel, wodurch der protektive, antiproliferative Einfluss des ER-beta abgeschwächt und die Kanzerogenese begünstigt wird.
Die Prostata als Speicher von Kanzerogenen
Im Laufe des Lebens können sich in der Prostata beträchtliche Mengen an Kanzerogenen ansammeln. Hierzu zählen klassische Kanzerogene, die beim Braten, Schmoren oder Grillen von Fleisch auftreten (z. B. heterozyklische Amine, polyzyklische aromatische Kohlenwasserstoffe), sowie exogene, östrogen wirksame Substanzen (z. B. PCB, Phthalate [verbreitete Weichmacher aus Kunststoff], Bisphenol A). Diese wirken einerseits direkt kanzerogen, und fördern andererseits durch ihre östrogene Wirkung die Proliferation. Auch Metalle wie Kupfer, Nickel und Eisen wirken proinflammatorisch, prooxidativ und dadurch prokanzerogen.
Viele Metalle wirken auch als Metalloöstrogene. Bei Metalloöstrogenen handelt es sich um anorganische Xenoöstrogene, die an Östrogenrezeptoren binden und die Genexpression in humanen, östrogensensitiven Zellen beeinflussen. Aufgrund ihrer Fähigkeit den Östrogenrezeptor zu aktivieren, werden Metalloöstrogene im Zusammenhang mit Brustkrebs diskutiert. Beispiele für Metalloöstrogene sind Aluminium, Arsen, Cadmium, Kupfer, Blei, Quecksilber und Nickel (Darbre, 2006).
Veränderungen der Biotransformation
Auch die Aktivität der Metabolisierungssysteme ist bei der Entstehung des Prostatakarzinoms von Bedeutung, denn Östrogene werden über Phase-1-Enzyme zu Kanzerogenen transformiert. Eine genetisch oder ernährungsbedingte Schwäche der Phase-2-Entgiftung bei gleichzeitiger Dominanz der Phase-1-Giftung ist ein entscheidender Schritt in Richtung Tumorentstehung, der sich mit steigendem Alter immer mehr auswirkt ‑ denn aus den im Alter zunehmend vorhandenen Östrogenen werden durch Phase-1-Enzyme krebserregende Chinone gebildet.
Zunehmende Fettspeicher und Aromataseaktivität
Der Hauptanteil männlicher Östrogene (Estradiol) wird aus Testosteron durch das Enzym Aromatase gebildet. Die Aromatase ist im Fettgewebe, aber auch direkt in der Prostata aktiv. Die Vermutung liegt nahe, dass der kanzerogene Effekt der Androgene in der Prostata zumindest teilweise über die Konversion von Testosteron zu Estradiol und über dessen Rezeptor ER-alpha erfolgt. Übergewicht, also eine Steigerung des Körperfettanteils und damit der Östrogenproduktion, ist zudem ein bekannter Risikofaktor für die Entwicklung verschiedenster Krebsarten. Sowohl für die Entwicklung der gutartigen Prostatahyperplasie als auch des Prostatakarzinoms spielt die Aromatase-Aktivität eine entscheidende Rolle (Hiramatsu et al., 1997; Matzkin und Soloway, 1992).
Bedeutung der Androgene für die Kanzerogenese
Androgene als Kanzerogene
Wachstum, Differenzierung und Funktion der Prostata stehen unter dem Einfluss der Androgene, deren Wirkung über den AR-Komplex vermittelt wird (Brinkmann et al., 1999; Cude et al., 1999; Cunha et al., 2004). Unter dem Einfluss der Steroide entfalten Wachstumsfaktoren, z. B. der Epidermal Growth Factor (EGF), zellproliferierende Eigenschaften auf die Prostata. Im Tierversuch wurde gezeigt, dass Androgene bei Ratten direkt Prostatakarzinome auslösen können (Bosland, 2000).
Das wichtigste auf genetischer Ebene agierende Androgen ist der Testosteron-Metabolit Dihydrotestosteron (DHT), der aus Testosteron durch die 5-alpha-Reduktase gebildet wird (s. Abb. 2): DHT moduliert die Expression von Genen, die mit Androgenen in Zusammenhang stehen (Griffiths et al., 1995). Die 5-alpha-Reduktase hat daher eine Schlüsselfunktion in der Entwicklung der Prostata. Die Höhe der 5-alpha-Reduktase-Aktivität, die zur DHT-Bildung führt, beeinflusst das Entstehungsrisiko eines Prostatakarzinoms (Ross et al., 1992; Eaton und Reeves, 1999). Umgekehrt reduziert die Hemmung dieses Enzyms die Konzentration an biologisch aktivem DHT. 5-alpha-Reduktase-Hemmstoffe werden therapeutisch in der Behandlung des Prostatakarzinoms eingesetzt.
Die potentiell kanzerogene Wirkung von Testosteron selbst ist möglicherweise darauf zurückzuführen, dass Testosteron bei Männern mengenmäßig der bedeutendste Vorläufer von Estradiol ist. Die Bedeutung von Estradiol für die Kanzerogenese wird im nächsten Abschnitt ausführlich besprochen. Im Gegensatz zu Testosteron kann DHT nicht in Estradiol überführt werden. Bei Ratten führte DHT nur bei 5 % der Tiere zu Karzinomen, die Kombination mit Estradiol jedoch erhöhte das Auftreten der Tumoren auf 15 % (Bosland, 2005).
Die Rolle von Testosteron wird kontrovers diskutiert. So tragen Testosteronpräparate eine Warnung, dass sie bei einem Prostatakarzinom nicht anzuwenden sind. Die Befürchtung, dass hohe Testosteronspiegel das Wachstum des Prostatakarzinoms fördern, stammt u. a. aus Beobachtungen, nach denen Eunuchen kein Prostatakarzinom entwickeln, sowie aus den Arbeiten von Huggins. Huggins und Hodges zeigten 1941 die therapeutische Wirkung einer Kastration von Männern mit fortgeschrittenem Prostatakarzinom und berichteten bei einem einzigen Patienten von einer schnellen klinischen Progression unter Testosterongabe (Huggins und Hodges, 1941; Morgenthaler, 2007). Später wurde jedoch belegt, dass sich solche negativen Effekte nur bei kastrierten Männern ergeben (Morgenthaler, 2008). Offizielle Studien zu Eunuchen und Prostatakarzinomen sind nicht auffindbar. Jedoch zeigte eine chinesische Studie (Wu und Gu, 1991), dass die Prostata von 26 Eunuchen durchschnittlich 54 Jahre nach Kastration entweder gar nicht oder nur sehr klein tastbar war.
Auch Imamoto et al. (2008) kommen in einem Review zu dem Schluss, dass die traditionelle Ansicht, höhere Testosteronspiegel würden ein höheres Risiko bedeuten, wenig faktische Evidenz hat. Eine Metaanalyse von acht epidemiologischen prospektiven Studien ergab keine signifikanten Unterschiede zwischen den Androgenspiegeln (einschließlich DHT und Testosteron) und Östrogenspiegeln von Männern, die in der Folge an Prostatakrebs erkrankten, und der Kontrollgruppe (Eaton et al., 1999).
Eine Untersuchung an 17.049 Männern brachte ein erstaunliches Ergebnis: Das Risiko für ein aggressives Prostatakarzinom wurde bei doppelten Testosteron- und Androstendion-Blutspiegeln (Nebennieren-Vorläufer von Androgenen und Östrogenen) fast halbiert und war bei der doppelten Konzentration von DHEA-Sulfat um 37 % niedriger. Ähnliche negative, aber statistisch nicht signifikante, lineare Trends wurden für freies Testosteron, Estradiol und Sexualhormon-bindendes Globulin (SHBG) beobachtet (Severi et al., 2006). Für das nichtaggressive Prostatakarzinom wurde im Zusammenhang mit den Blutwerten dieser Hormone keine Risikoerhöhung festgestellt. Diese Ergebnisse geben Sinn wenn man bedenkt, dass es beim Prostatakarzinom auf das Zusammenspiel von Hormonen und Rezeptoren ankommt: Hohe Hormonspiegel führen zu einer Herabregulierung der Rezeptoren und umgekehrt.
Bei einer weiteren Untersuchung an 326 Prostatakrebspatienten wiesen niedrige präoperative Testosteronwerte eine Korrelation mit einem fortgeschritteneren pathologischen Stadium auf (Isom-Batz et al., 2005). Auch eine Studie mit 2254 Patienten stützt die Hypothese, dass niedriges freies Serum-Testosteron ein Marker für einen aggressiveren Prostatakrebs sein kann, besonders bei Patienten mit einem PSA-Wert von ≤ 4 ng/ml (D’Amico et al., 2002).
Bei 96 Patienten mit Prostatakrebs und Knochenmetastasen waren unter Hormontherapie hohe Testosteronwerte und niedrige LH- (luteinisierendes Hormon), FSH- (follikelstimulierendes Hormon) und Prolaktinwerte günstige prognostische Faktoren, unabhängig vom Tumorgrading (Chen et al., 2002).
Dennoch ist eine Testosteron-Supplementierung sicher keine generelle Antiaging-Therapie. In einer Studie von Gaylis et al. (2005) wurde bei 20 Patienten die Entstehung von Prostatakrebs mit Testosteron-Supplementierung in Verbindung gebracht. Insbesondere bei kastrierten Männern, wo unter Androgenentzug der AR häufig hypersensitiv, überexprimiert oder mutiert ist, könnte eine Testosteron-Supplementierung zu einer massiven Tumorproliferation führen – analog zu der bereits erwähnten Beobachtung von Huggins und Hodges (1941).
Androgenrezeptor-Expression beim Prostatakarzinom
Der AR spielt in der Kanzerogenese vermutlich eine zentrale Rolle. Dies untermauert ein Tierversuch, bei dem ein AR-Antagonist die Entwicklung eines Prostatakarzinoms durch ein Kanzerogen nahezu komplett unterbinden konnte (unveröffentlichte Studienergebnisse, erwähnt in: Bosland, 2005).
Der AR ist im inaktiven Stadium an Hitzeschock-Proteine (HSP) gebunden. Erst durch die Bindung von DHT erfolgen die Dissoziation von HSP und die DNA-Bindung, was die Aktivierung der Androgen-Zielgene bewirkt und so zu Proliferation, Zellerhalt, PSA-Sekretion etc. führt. Insbesondere unter Hormonentzug verändert sich die AR-Situation, v. a. durch Hyperexpression/-sensitivität und Mutation (Röpke et al., 2004).
Die AR-Expression der Prostatakrebszellen ist unterschiedlich und hängt vom Tumorstadium ab. Im gewöhnlichen, hormonsensitiven Prostatakarzinom wird die Hauptmasse von exokrinen Tumorzellen gebildet, die zellbiologische Ähnlichkeiten mit dem sekretorischen Zelltyp im Differenzierungskompartiment des gesunden Prostataepithels aufweisen. Durch Androgenentzug können sie in die Apoptose getrieben werden (hormonsensitiv).
Im Androgen-insensitiven Tumorstadium ist der AR paradoxerweise häufig überexprimiert. Dadurch werden die Tumorzellen hypersensitiv gegenüber den restlichen Androgenen nach Androgenentzug, der keine Apoptose mehr auslöst (hormonrefraktär). Zellbiologisch ähneln die Tumorzellen in diesem Stadium den Basalzellen des normalen Prostataepithels.
Im fortgeschrittenen und Androgen-insensitiven Stadium werden zudem „basalzellspezifische“ Gene wieder exprimiert, deren Expression in früheren Stadien herunterreguliert ist. Demnach nimmt das Androgen-insensitive Prostatakarzinom zellbiologische Eigenschaften der Basalzellen bzw. Prostatastammzellen wieder auf. Der Hauptunterschied zwischen Basalzellen und hormonrefraktären Prostatakarzinomzellen ist die starke Überexpression des AR, die als Gegenregulation auf den Androgenentzug zu verstehen ist (Li et al., 2004a; Bonkhoff und Remberger, 1996).
Mutationen im AR-Gen werden vor allem bei metastasierten oder hormonrefraktären Prostatakarzinomen beschrieben. Verschiedene Steroidhormone weisen eine höhere Affinität zu diesem mutierten AR auf, führen dadurch zu einer höheren transkriptionellen Aktivität und stimulieren die Proliferation (Röpke et al., 2004).
Eine Studie von Montgomery et al. (2008) zeigt, dass metastasierte Prostatakarzinome kastrierter Männer verstärkt Enzyme exprimierten, die für die Synthese von Testosteron und DHT aus Cholesterin nötig sind. So konnten die Tumoren Androgenspiegel aufrechterhalten und AR-Zielgene aktivieren, um ihr Überleben zu sichern. Auf diese Weise können Karzinome trotz sehr niedriger Androgen-Blutwerte überleben.
Dies zeigt einerseits, dass auch Androgen-unabhängige Karzinome nicht wirklich Androgen-unabhängig sind, sondern sich einfach von der fehlenden externen Androgenzufuhr (bei Androgen-Entzugstherapie) unabhängig gemacht haben. Andererseits wird deutlich, wie jeder extreme Eingriff zu einer Gegenregulation führt. Je niedriger die Androgenspiegel im Blut auf Dauer sind, desto mehr wird die Entwicklung der „scheinbar“ Androgen-unabhängigen, aggressiveren Karzinome forciert.
Neuroendokrine Differenzierung im Prostatakarzinom
Bonkhoff und Fixemer (2004) beschreiben die neuroendokrine Differenzierung im Prostatakarzinom als einen unerkannten und therapierefraktären Phänotyp: Neuroendokrine Zellen (Marker: Chromogranin A; ChrA) sind primär Androgen-insensitiv, da sie keinen AR tragen. Zudem sind neuroendokrine Tumorzellen gegen konventionelle Strahlentherapie resistent. Die Hauptmasse der ChrA-positiven Tumorzellen ist daher potentiell unsterblich und somit therapierefraktär. Das Ausmaß der neuroendokrinen Differenzierung (messbar an der ChrA-Expression) nimmt im Rahmen der Tumorprogression und der Entstehung der Androgenresistenz kontinuierlich zu. Die höchsten ChrA-Expressionsraten und Serumwerte finden sich bei Patienten mit klinisch Androgen-insensitiven Karzinomen. Neuroendokrine Krebszellen haben eine langsame Proliferation, nähren jedoch durch Wachstumsfaktoren exokrine Tumoren in der Umgebung. Neuroendokrine Karzinome können sich unter Hormonblockade nach mehreren Jahren entwickeln, sind besonders aggressiv und gehen nicht mit einem wesentlichen PSA-Anstieg einher. Unter Hormonblockade können sich auch aus ehemals PSA-positiven Karzinomzellen neuroendokrine Karzinomzellen entwickeln.
Tabelle: Grobeinteilung der Prostatakarzinomzelltypen
| Krebszelltyp | Marker | Hormonstatus | Prognose | Therapie |
| Hormonsensitiv | Ähnlichkeiten mit sekretorischem Zelltyp des gesunden Prostataepithels (Zytokeratinmuster, PSA-Produktion, Androgenrezeptor) | Androgen-abhängig | Gut (in Abhängigkeit von Gleason-Score und Metastasierung) | Stadien-abhängig:active surveillance/ watchful waiting,Prostatektomie,BestrahlungHormonblockade |
| Stammzell- artig | Ähnlichkeiten mit Basalzellen des gesunden Prostataepithels (Expression von z. B. erbB-2, erbB-3, HER2-neu, EGFR und Bcl-2) | Scheinbar Androgen-unabhängig;Starke Hypersensitivität und/oder Über-expression des Androgenrezeptors;Eigensynthese von Androgenen | Schlecht | OperationOff Label |
| Neuro-endokrin | Chromogranin A | Echt Androgen-unabhängig | Schlecht | Operation |
Bedeutung der Östrogene für die Kanzerogenese
Einfluss des Östrogenspiegels
Auch wenn das Prostatakarzinom ein Androgen-abhängiger Tumor ist, ist die Bedeutung der Östrogene vielfach dokumentiert (Bosland, 2000). Ihre Kanzerogenität auf die Prostata ist im Tiermodell an Ratten belegt. Dabei potenzieren sie die kanzerogenen Eigenschaften des Testosterons: Ratten entwickeln unter Langzeitbehandlung mit Testosteron und Estradiol oder Diethylstilbestrol in 100 % der Fälle ein Prostatakarzinom (Bosland et al., 1995).
Epidemiologische Studien legen einen direkten Zusammenhang zwischen dem Östrogenspiegel und dem Prostatakrebsrisiko nahe. Schwarze US-Amerikaner haben höhere Serum-Estradiol-Werte als Amerikaner kaukasischer Abstammung und mit einer Inzidenz von 181 pro 100.000 Einwohnern eine fast doppelt so hohe Inzidenz von Prostatakarzinomen (Rohrmann et al., 2007). Im Gegensatz dazu haben Japaner sowohl besonders niedrige Östrogenspiegel im Blut als auch eine besonders niedrige Prostatakarzinominzidenz (6,7 pro 100.000) (De Jong et al., 1991). Die bereits erwähnte Untersuchung an einer Kohorte von 17.049 Männern ergab keinen Zusammenhang zwischen Estradiol und einem Prostatakarzinom. (Severi et al., 2006). Entscheidend dürften daher nicht nur Blutspiegel, sondern die individuellen Verhältnisse von Androgenen, Östrogenen, alimentär aufgenommenen Phytoöstrogenen und der Rezeptorensituation sein. Beim älteren Mann, bei dem auch das Prostatakrebsrisiko stark zunimmt, verschiebt sich physiologischerweise das Gleichgewicht von Androgenen und Östrogenen um bis zu 40 % zugunsten der Östrogene (Ho et al., 2006b).
Einfluss der Biotransformation – Giftung von Östrogenen zu Kanzerogenen
Phase-1-Enzyme der Cytochrom-P450-Superfamilie transformieren Östrogene zu 2-Hydroxy- und 4-Hydroxy-Catechol-Östrogenen und ihren Semi-/Chinonen, welche zu den potenten genotoxischen Kanzerogenen zählen. Semichinone entfalten ihre Genotoxizität durch ihren Radikalcharakter und verursachen zusätzlichen oxidativen Stress (Cavalieri et al., 2000; Jefcoate et al., 2000), Chinone reagieren mit DNA-Basen zu DNA-Addukten. Durch Phase-2-Enzyme wie beispielsweise die Catechol-O-Methyltransferase (COMT) werden diese genotoxischen Östrogen-Metaboliten entgiftet. Das Phase-2-Enzym Glutathion-S-Transferase entgiftet Chinone, fängt Semichinon- und weitere freie Radikale ab und begrenzt dadurch das Ausmaß der oxidativen Zellschäden.
Eine Reihe experimenteller Befunde belegt, dass die individuelle Enzymausstattung, insbesondere das Gleichgewicht von Phase-1- zu Phase-2-Enzymen für die Pathogenese von Tumoren entscheidend ist. Dieses Enzymgleichgewicht wird einerseits auf genetischer Ebene reguliert (Expression), kann andererseits aber auch über die Nahrung beeinflusst werden (Aktivität). Daher sind für die Chemoprävention Pflanzenstoffe wichtig, welche Phase-2-Enzyme aktivieren und Phase-1-Enzyme hemmen. Diese Effekte sind für Flavonoide bereits bekannt und wurden ebenfalls für Granatapfel-Polyphenole vermutet und z. T. nachgewiesen.
Zwei unterschiedliche Östrogenrezeptoren
Die Wirkung von Östrogenen wird über die beiden Östrogenrezeptoren ER-alpha und -beta vermittelt. Bis Mitte der 1990er Jahre war lediglich der ER-alpha bekannt, bis 1996 Jan-Ǻke Gustafsson den ER-beta entdeckte, der sich in Struktur, Wirkung und Gewebsverteilung deutlich von ER-alpha unterscheidet.
In der Leber und Gebärmutter überwiegt ER-alpha; in Knochen, Darm, Gefäßwänden sowie der Prostata ER-beta. In Mammae, Ovarien und Gehirn sind beide Subtypen in etwa gleichgewichtig. Die Östrogenrezeptoren gehören zu der Klasse der nukleären Hormonrezeptoren und besitzen sechs Domänen (A-F). Deutliche Unterschiede zwischen den beiden Subtypen ER-alpha und ER-beta liegen in der A/B-Domäne (ligandenunabhängige transkriptionelle Aktivierungsfunktion) und der F-Domäne (Liganden-Bindungsdomäne) (Kuiper et al., 1996), was zu sehr unterschiedlichen biologischen Effekten führt. Während das endogene Estradiol und Estron an beide Rezeptoren etwa gleichermaßen binden, haben pflanzliche Phytoöstrogene in der Regel zu ER-beta die höhere Affinität.
Auch innerhalb der Prostata unterscheiden sich die Expressionsmuster in den Gewebstypen und die biologischen Funktionen der ER-Subtypen deutlich. Im sekretorischen Epithel überwiegt ER-beta, während ER-alpha vor allem im Stroma und in geringerem Maße in der Basalzellschicht exprimiert wird. Im Stroma bewirken Östrogene über ER-alpha die Freisetzung von Wachstumsfaktoren, welche zu einer Proliferation des Epithels führen (Imamov et al., 2005). So kommen dem ER-alpha wachstums- und proliferationsfördernde Aufgaben zu, während der ER-beta proliferationshemmende und differenzierende Wirkungen vermittelt.
Die Bedeutung des Östrogen-Rezeptors alpha in der Prostata
Der ER-alpha ist der wichtigste Östrogenrezeptor in Basalzellschicht und Stroma, wo er die Synthese von Wachstumsfaktoren stimuliert. Bei der malignen Transformation des Prostataepithels verlagert sich die Expression des ER-alpha auf mRNA-Ebene konstant in das sekretorische Epithel (Bonkhoff et al., 1999). Unter den verschiedenen durch ER-alpha-regulierten Genen ist der Progesteron-Rezeptor (PR) einer der wichtigsten Marker der östrogenregulierten Zellproliferation in hormonabhängigen Tumoren. Die Expression des PR verläuft im Prostatakarzinom parallel zu der von ER-alpha (Bonkhoff et al., 2001). Am ausgeprägtesten ist die Expression von PR in hormonrefraktären und metastasierten Prostatakarzinomen.
Im Gegensatz zum Mammakarzinom ist die verstärkte Expression von ER-alpha und PR im Epithel ein späteres Ereignis in der Tumorprogression des Prostatakarzinoms und korreliert mit dem Gleason-Grad und dem pathologischen Stadium. Demnach wirkt der ER-alpha als Onkogen, das bei der malignen Transformation des Prostataepithels überexprimiert wird und somit den kanzerogenen Effekt der Östrogene auf das sekretorische Prostataepithel vermitteln kann (Bonkhoff et al., 1999). Die Blockierung des ER-alpha ist ein rationaler Ansatz für die Chemoprävention des Prostatakarzinoms. Dafür sprechen auch erste klinische Ergebnisse (z. B. Price et al., 2006) über die präventive Wirkung des ER-alpha-Antagonisten Toremifen. Auch Granatapfel-Polyphenole zeigen eine ER-alpha-antagonistische Wirkung.
Die Expression von ER-alpha und -beta kann durch verschiedene östrogene Substanzen beeinflusst werden. So steigern Östrogene die Expression von ER-alpha und senken die ER-beta-Expression. Dies fördert die Entwicklung von hyper-, dys- und neoplastischen Läsionen der adulten Prostata (Ho et al., 2006a). Dagegen steigerten im Uterus von ovarektomierten Mäusen der ER-beta-Agonist Genistein und SERMs (selective estrogen receptor modulator) wie Tamoxifen und Toremifen die Expression von ER-beta. Genistein und das Antiöstrogen ICI (Faslodex) senkten zusätzlich die Expression von ER-alpha (Wu et al., 2007).
Die ER-Situation scheint also auch von externen Stimuli abhängig zu sein und ist über ER-alpha in eine proliferative oder über ER-beta in eine antiproliferative Richtung dirigierbar. Bei der heutigen westlichen Ernährungsweise und bei Übergewicht (hoher Körperfettanteil begünstigt die Estradiol-Biosynthese) überwiegen hochwirksame Östrogene wie 17-beta-Estradiol, die den ER-alpha stimulieren und damit die Proliferation des Prostataepithels fördern.
Die Bedeutung des Östrogen-Rezeptors beta in der Prostata
Der ER-beta existiert in fünf Isoformen (ER-beta 1-5), die unterschiedliche Funktionen und Gewebsverteilungsmuster besitzen. ER-beta 1 ist die einzige funktionstüchtige Isoform und kann als einzige Isoform Homodimere bilden. Für ER-beta 2, 4 und 5 wurde gezeigt, dass sie mit ER-beta 1 Heterodimere bilden und dadurch die Signaltransduktion von ER-beta 1 verstärken können. Dabei hängen die verstärkenden Effekte vom Liganden ab: Estradiol und synthetische Östrogene (z. B. Diethylstilbestrol, Bisphenol A) induzieren die Homodimerisierung von ER-beta 1 sowie die Heterodimerisierung mit ER-beta 2, 4 oder 5, während Phytoöstrogene (z. B. Genistein, Apigenin) lediglich ER-beta-1-Homodimere mit geringerer östrogener Transaktivierungsaktivität induzieren (Leung et al., 2006).
In hormonrefraktären PC3-Prostatakarzinomzellen wurden hohe Werte für die ER-beta-2-Isoform, in hormonrefraktären DU145-Prostatakarzinomzellen extrem hohe Werte für die ER-beta-5-Isoform gemessen. Bei allen Karzinomzelllinien war die Konzentration der ER-beta-1-Isoform, die als einzige klassische Homodimere mit geringerer östrogener Transaktivierungsaktivität bilden kann, gering. Dies legt nahe, dass es in Prostatakarzinomzellen verstärkt zu einer Bildung von ER-beta-Heterodimeren kommt, die eine höhere östrogene Transaktivierungsaktivität haben als die Homodimere (Leung et al., 2006). Möglicherweise ist eine Verschiebung des Verteilungsmusters der ER-beta-Isoformen der Grund, warum in Prostatakarzinomzellen die Tumorsuppressorfunktion von ER-beta zurückzugehen scheint.
Bisher wird ER-beta in Studien meistens als „ein“ Rezeptor behandelt und die wahrscheinlich unterschiedlichen Auswirkungen der verschiedenen Isoformen und ihrer Heterodimere nicht berücksichtigt. Diese Überlegungen vorab sollen folgende Texte zur allgemeinen und prostataspezifischen Wirkung von ER-beta etwas relativieren.
Selektive ER-beta-Agonisten zeigten im Tiermodell antiinflammatorische Effekte bei chronisch entzündlichen Darmerkrankungen, Arthritis und Endometriose (Harris, 2006). Entzündungsprozesse spielen sowohl in der Kanzerogenese als auch bei der Entstehung von Alterserkrankungen und Arteriosklerose eine wichtige Rolle. Der ER-beta vermittelt im Tier- und Zellversuch bei Brustkrebs und in der Prostata antiproliferative und antientzündliche Effekte (Koehler et al., 2005). Diese Ergebnisse zeigen, dass der ER-beta sowohl in der Prävention als auch in der Therapie von Alters-, Herz-Kreislauf- und Krebserkrankungen von Bedeutung ist (Koehler et al., 2005).
Der ER-beta ist in die Bildung antioxidativer Schutzenzyme involviert und schützt damit indirekt vor oxidativem Stress. Antioxidative Schutzenzyme, wie die Phase-2-Enzyme Chinonreduktase und Glutathion-S-Transferase, spielen bei der Entgiftung von freien Radikalen und Chinonen, die bei Phase-1-Reaktionen aus Substraten wie z. B. Estradiolmetaboliten entstehen, eine wichtige Rolle. Für die Expression der Chinonreduktase wurde eine deutliche Abhängigkeit von der Stimulation des ER-beta gezeigt – die Aktivierung von ER-alpha führte zu deutlich geringeren Expressionsraten des Enzyms (Montano et al., 2000). Ähnliche Effekte zeigten sich für die Expression der Glutathion-S-Transferase (Montano et al., 2004). In Brustkrebszellen reduzieren ER-beta-selektive Phytoöstrogene Estradiol-induzierte oxidative DNA-Schäden, wobei gleichzeitig eine ER-beta-abhängige Steigerung der Chinonreduktase-Expression festgestellt wurde (Bianco et al., 2005).
Während ER-alpha Proliferation und Wachstum unterstützt und damit die Entstehung hormonabhängiger Tumoren fördert, vermittelt ER-beta die Zelldifferenzierung und schützt damit vor Tumoren. Der ER-beta vermittelt seine protektiven Wirkungen dadurch, dass er die Signaltransduktion (Lindberg et al., 2003), aber auch die Expression von ER-alpha moduliert (Imhof et al., 2006).
Vieles deutet darauf hin, dass ER-beta in der Prostata der wesentliche ER ist, als Tumorsuppressor agiert und eine antiproliferative, differenzierende Schutzwirkung hat (Imamov et al., 2005). Ein ER-beta-knockout Experiment an Mäusen verdeutlicht dies: Wurde den Mutanten mit Prostatakarzinom das ER-beta-Gen mittels Adenoviren wieder zugefügt, gingen Invasion und Wachstum der Prostatakarzinomzellen zurück. In Zellen, die ER-beta im Übermaß exprimierten, wurde Apoptose ausgelöst, während die Prostata von ER-beta-knockout Mäusen eine verminderte Apoptoserate zeigte (Cheng et al., 2004).
Der ER-beta ist im gesunden Prostataepithel ein klarer Tumorsuppressor, doch möglicherweise verändert sich diese Rolle im Prostatakarzinom – entweder durch verminderte ER-beta-Expression, oder indem seine Tumorsuppressorfunktion zurückgeht (Fixemer et al., 2003). Bonkhoff und Mitarbeiter gehen davon aus, dass ER-alpha und ER-beta in hormonrefraktären Prostatakarzinomen Heterodimere bilden (Fixemer et al., 2003), wobei der proliferative ER-alpha der dominierende Aktionspartner zu sein scheint. Dies wird durch molekularbiologische Untersuchungen bestätigt (Li et al., 2004b) und ist ein Hinweis darauf, dass im hormonrefraktären Stadium der ER-beta durch Heterodimerbildung mit ER-alpha seine antiproliferative Wirkung verliert und die proliferative Wirkung von ER-alpha dominiert.
Untersuchungen an der unreifen Prostata von Mäusen legen nahe, dass auch zwischen der Expression von ER-beta und dem AR eine Beziehung besteht (Weihua et al., 2002): So weisen proliferierende Zellen in der unreifen ventralen Prostata deutlich höhere Konzentrationen des AR bei gleichzeitig geringer ER-beta-Expression auf, während das Verhältnis bei nicht proliferierenden Zellen umgekehrt ist. In Abwesenheit des ER-beta kommt es vermutlich entweder zu einer Steigerung der mRNA des AR oder zu einer Verminderung seines Abbaus.
3beta-Adiol ist der wohl wichtigste endogene ER-beta-Agonist und spielt bei der Regulation des Prostataepithels eine wichtige Rolle. Es wird in der Prostata aus DHT unter Einwirkung des Enzyms 3beta-Hydroxysteroiddehydrogenase (17 HSD 3) gebildet (s. Abb. 2). Der Androgenmetabolit erreicht in der gesunden menschlichen Prostata deutlich höhere Konzentrationen als Estradiol (Voigt und Bartsch, 1986). Im Gegensatz zu Estradiol, das mit gleichstarker Affinität an ER-alpha und -beta bindet, zeigt das schwächer affine 3beta-Adiol eine leichte Bindungsselektivität zum ER-beta-Rezeptor. Viel größer ist jedoch die Selektivität bezüglich der intrinsischen Aktivität: Im ERE-Luziferase-Assay (ERE = estrogen responsive element) steigerte 3beta-Adiol bei gleicher Konzentration die ERE-Promotor-Aktivität von ER-beta stärker als das viel höher affine Estradiol. Trotz niedrigerer Affinität zeigte 3beta-Adiol an ER-beta also eine höhere Wirkung als Estradiol. 3beta-Adiol bindet und blockiert zudem den ER-alpha, zeigt aber in Bezug auf die ERE-Promotor-Aktivität von ER-alpha eine extrem geringe Wirkung (Pak et al., 2005). Dies bedeutet für das Wirkprofil von 3beta-Adiol, dass es sich schwerpunktmäßig um einen ER-beta-Agonisten und um einen ER-alpha-Antagonisten handelt und damit einen bedeutenden endogenen, antiproliferativen Schutzfaktor darstellt. Eine Studie lieferte zudem einen Hinweis darauf, dass 3beta-Adiol vermutlich als Komplex mit ER-beta bei der Regulation der AR-Expression eine große Rolle spielt und überschießendes epitheliales Wachstum verhindert (Weihua et al., 2001).
Letztlich ist das Zusammenspiel zwischen ER-alpha, ER-beta, PR, AR und ihren Isoformen, Liganden und Coaktivatoren sehr komplex. Der Forschungsbedarf ist noch groß, insbesondere auch in Bezug auf die ER-beta-Isoformen und deren sich veränderndes Verteilungsmuster und Wirkungsweise während der Kanzerogenese. Fraglich ist, ob sich ein solch komplexes und hochgradig dynamisches Zusammenspiel letztlich so aufklären lässt, dass sich daraus für den Patienten und seine individuelle, ebenso komplexe Tumorbiologie klare Konsequenzen ergeben. Allgemeingültige Antworten wird es wohl letztlich nicht geben, denn kein Betroffener und kein Prostatakarzinom gleicht dem anderen.
Phytoöstrogene wie Soja-Isoflavone können Prostatakrebs und möglicherweise auch der gutartigen Prostatahyperplasie vorbeugen. Auch bei einem normalen Prostatakarzinom wirken sie günstig.
Als Arzt und Betroffener sollte man immer daran denken, dass sich beim fortgeschrittenen, metastasierten Prostatakarzinom vor allem unter Anti-Hormontherapie die Tumorbiologie sehr dynamisch verändert und normalerweise gesunde Stoffe, wie z. B. Vitamin D (Ahn et al., 2008) oder Isoflavone (Kurahashi et al., 2007), u. U. eine paradoxe Wirkung haben können, indem sie das Tumorwachstum fördern. So wie sich die individuelle Beschaffenheit der Tumorbiologie im Krankheitsverlauf allmählich verändert, muss sich auch die Therapie anpassen. Daher sollte der Erfolg der Naturstoffe, insbesondere auch hochdosierter Phytoöstrogene, und auch der Erfolg von Änderungen in der Einnahme an der PSA-Dynamik gemessen werden.
Prooxidative und proinflammatorische Wirkung von Kupfer und Eisen
Auch Kupfer spielt möglicherweise eine bislang weitgehend unterschätzte Rolle in der Kanzerogenese. Das Metall wirkt zytotoxisch und erzeugt oxidativen Stress und chronische Entzündungsreaktionen durch Interaktionen mit NF-kappaB und TNF-alpha (Kennedy et al., 1998; Persichini et al., 2006). Zudem stimuliert es die Neubildung von Tumorgefäßen. Die Kupferwerte von Krebskranken sind gegenüber Gesunden deutlich erhöht (Nayak Shivananda et al., 2003). In einer Untersuchung an 3000 Männern und 3244 Frauen erhöhten die prooxidativen Metalle Eisen und Kupfer das relative Krebserkrankungsrisiko um den Faktor 1,86 (Eisen) bzw. 1,89 (Kupfer), während niedrige Serumwerte das relative Risiko auf 0,96 (Eisen) bzw. 0,76 (Kupfer) senkten (Wu et al., 2004). Insbesondere innerhalb von vier Jahren vor Diagnosestellung eines Karzinoms stiegen die Kupferserumwerte, was ein Hinweis darauf sein könnte, dass die Erhöhung der Kupferserumwerte mit dem Tumorwachstum in Zusammenhang steht oder durch den Tumor verursacht wird (Coates et al., 1989).
Therapeutisch verhindern Chelatoren die Kupferaufnahme und reduzieren deutlich das Krebswachstum und die Neoangiogenese (Brewer et al., 2000). Insbesondere bei der Entstehung von Alterskarzinomen wie dem Prostatakarzinom könnte die weit verbreitete, chronische Kupferexposition eine bedeutende Rolle spielen. In Deutschland sind meist Kupferleitungen im Haushalt die wichtigste Quelle für eine Kupferüberversorgung.